Turner Sendromu Hakkında Bilmeniz Gereken Her Şey


tarafından incelendi ve onaylandı. doktor José Gerardo Rosciano Paganelli
Turner Sendromu cinsiyet kromozomlarını etkileyen bir genetik bozukluktur. Bir X kromozomunun tamamen ya da kısmen olmayışıyla nitelendirilir, bu yüzden sadece kadınları etkiler. Karyotip bu nedenle 45 X olacaktır.
Doğal olarak, tek bir X kromozomunun varlığı, birincil ve ikincil kadın cinsel özelliklerinin gelişimini etkiler.
İstatistikler
İstatiksel olarak Turner sendromu vakalarının %50 sinde ikinci kromozomun tamamen kaybedildiği 45 X karyotip bulunur. Diğer yüzde elli ise sadece kısmi kromozom kaybı vardır. Bu durumlarda, kromozomun uzun ya da kısa kolu etkilenebilir.
Kalan durumlar mozaisizmlerdir. Bunlar genetik bozukluklar ile iki veya daha fazla hücresel çizginin olmaması ile nitelendirilir. Böylece, bu insanlar genetik olarak normal olan bazı hücrelere (45 XX) ve genetik olarak bozuk olan bazı hücrelere (45 X) sahip olacaklardır.
Normal durumlar altında cinsiyet kromozomları
Normal durumlar altında herkes 23 çift kromozoma sahiptir. Bunlardan 22 tanesi cinsiyet dışı otozoma veya kromozoma, ve final çifti ise cinsiyet çiftine karşılık gelir.
Cinsiyet kromozomlarının biri babadan, biri anneden gelir ve bebeğin genetik cinsiyetini belirler.
- İki X kromozomu olması (46 XX) cinsiyetin dişi olacağını belirler. Böylece birincil olarak kadın ve ikincil bir cinsiyet özellikleri gelişir.
- Bir Y kromozomu olması (46 XY) cinsiyetin erkek olacağını belirler. Böylece birincil olarak eril ve ikincil bir cinsiyet özellikleri gelişir.
Turner Sendromunda ne olur?
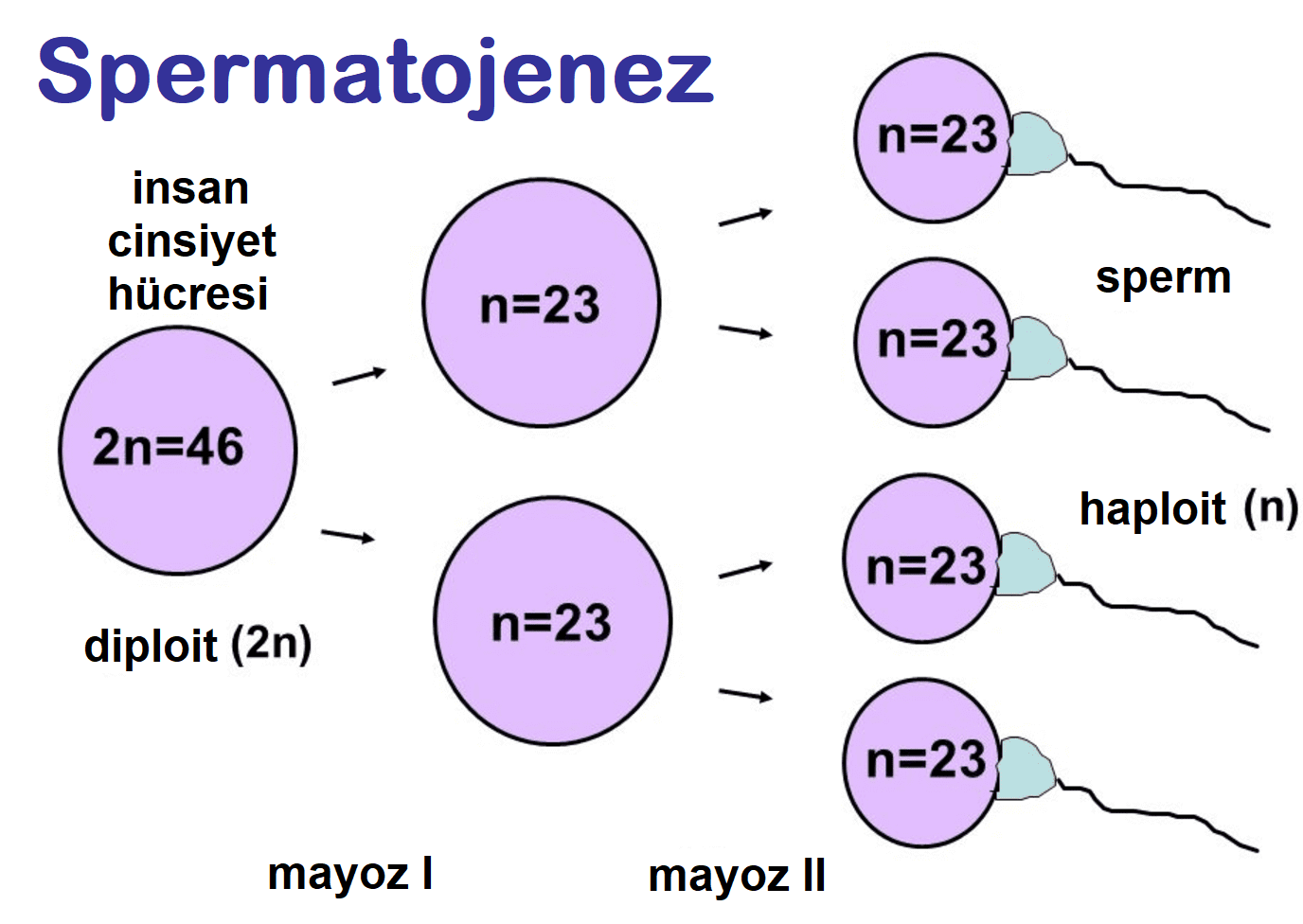
Bu kromozom bozukluğunu açıklanmadan önce, üreme hücresinin bölünmesinin (gametlerin) belirli yönlerini hatırlamak önemlidir.
Bu süreç mayoz olarak bilinir, mayoz I ve mayoz II olan 2 kısımdan oluşur. Bu sürecin yegane amacı ata hücreden gelen yarım genetik bilgiyi taşıyan çocuk hücreler üretmektir.
- Basitçe, 23 çift kromozomu olan kök hücre ile başlanır.
- Mayoz I sonunda, her birinde 23 kromozom bulunan 2 çocuk hücre oluşur.
- Mayoz II sonunda, bu iki hücre tekrar 23 kromozomlu 2 hücre daha üretir.
- Sonuç olarak, bir diploid kök hücre dört tane haploid çocuk hücre üretir.
Bir hücre nasıl kromozom kaybeder, ve bu durumda ne olur?
Genel olarak kabul gören teori, ikinci X veya Y kromozomunun gebelikten sonra kaybolduğudur. Yani, hücre bölümündeki bir hatadan kaynaklanan bir sorundur.
Y Kromozomunun olması dişi cinsiyet özelliklerinin gelişmesini belirler. Orijinal hücre XY olsa bile, bu kayıp dişinin gelişmesine sebep olur.
Turner Sendromunun klinik belirtileri

Fiziksel görünüş
- Çocukluk döneminde büyümede gecikme ve yetişkinlik döneminde boy kısalığı.
- Düzleştirilmiş burun kemeri, alçak göz çukurları ve göz kapaklarının hafif kusurlu olması gibi “Sfenks” benzeri yüz özellikleri.
- Normalden kısa ve geniş ense, çok alçak saç çizgisi.
- Çok sayıda renkli ben.
- Geniş göğüs kafesi, az gelişmiş çocuksu görünüme sahip ayrık meme uçları.
- İkincil cinsiyet özellikleri çok az gelişmiştir ve cinsel çocuksuluk vardır. Memeler gelişmemiştir, cinsel bölgelerde kıllanma olmaz ve yuvarlak kalçaya sahiptirler. Bütün bunlar, östrojen eksikliğinden kaynaklanır.
- Kolların dışarı dönük olması gibi kemik kusurları çok yaygındır.
Bu makaleyi ziyaret edin: Güçlü Kadınlara Özgü Nitelikler
İç bozukluklar
Ne yazık ki bu durum birtakım iç bozuklukları da beraberinde getirir, bunlar:
- Ana atardamar daralması ve kalp kapakçığı bozuklukları gibi doğuştan olan doğuştan gelen kalp rahatsızlıkları çok yaygındır.
- Önemli sayıda hastada böbrek sorunları ortaya çıkar, bunlardan en yaygını at nalı şeklinde böbrektir.
- Gadonal farklılaşma. Bu durumda yumurtalıklar az gelişmiştir, ve bunlar yerine gadonal bantlar vardır. Bu durum hipergonadotropik hipogonadizme sebep olur, yani östrojen üretimi için gerekli bütün uyarıcılar olmasına rağmen, yumurtalıklar olmadığı için üretilemezler.
- Amenore (Menstural döngünün olmaması) ve kısırlık. hastaların sadece küçük bir yüzdesi çocuk sahibi olabilir. Bu Turner Sendromunun en yaygın özelliklerinden birisidir.
Teşhis

Turner Sendromunun teşhis edilmesi zor olabilir. Bu bozukluğun olduğu çoğu kadın belirgin özelliklere sahip değildir. Hafif dışyapıya sahiptirler, çünkü daha ciddi vakalar düşükle sonuçlanır.
Vakaların üçte biri bebekken teşhis edilir. Belirli fiziksel özellikler ve kalp hastalığı belirtileri şüphe uyandıran belirtilerdir. Diğer üçte bir ise kısa boylarından dolayı çocukken teşhis edilir.
Son olarak, son üçte birlik kısım ise büyüme çağında teşhis edilir. Bu durumlarda, cinsel çocuksuluk ile birlikte kısa boy açıkça gözlemlenebilir ve hasta asla ergenliğe erişemez.
Tedavi
Östrojen kullanarak yapılan hormon replasmanı tedavisi (Hormone replacement therapy) cinsel özelliklerin gelişmesine yardımcı olur fakat kısırlığı tedavi edemez. Çocukluk döneminde, kız çocuklarında boy uzunluğunu normal seviyelere getirebilmek için büyüme hormonuyla tedavi tavsiye edilir.
Çocuk sahibi olabilirler mi?
Evet, Turner Sendromuna sahip kadınların çocuk sahibi olmaları mümkündür. Turner Sendromuna sahip insanların küçük bir yüzdesi çocuk sahibi olabilir ve gebe kalmaları için bir problem yoktur.
En sık rastlanan durum olan kısırlıkta ise, bağışlanmış yumurtalar ile in-vitro fertilizasyon (tüp bebek) tekniği kullanılmaktadır. Yumurta yapay ortamda döllenerek hastaya aşılanır. Bu gebelikler normal gebeliklere göre çok daha sıkı kontrol gerektirmektedir.
Turner Sendromu cinsiyet kromozomlarını etkileyen bir genetik bozukluktur. Bir X kromozomunun tamamen ya da kısmen olmayışıyla nitelendirilir, bu yüzden sadece kadınları etkiler. Karyotip bu nedenle 45 X olacaktır.
Doğal olarak, tek bir X kromozomunun varlığı, birincil ve ikincil kadın cinsel özelliklerinin gelişimini etkiler.
İstatistikler
İstatiksel olarak Turner sendromu vakalarının %50 sinde ikinci kromozomun tamamen kaybedildiği 45 X karyotip bulunur. Diğer yüzde elli ise sadece kısmi kromozom kaybı vardır. Bu durumlarda, kromozomun uzun ya da kısa kolu etkilenebilir.
Kalan durumlar mozaisizmlerdir. Bunlar genetik bozukluklar ile iki veya daha fazla hücresel çizginin olmaması ile nitelendirilir. Böylece, bu insanlar genetik olarak normal olan bazı hücrelere (45 XX) ve genetik olarak bozuk olan bazı hücrelere (45 X) sahip olacaklardır.
Normal durumlar altında cinsiyet kromozomları
Normal durumlar altında herkes 23 çift kromozoma sahiptir. Bunlardan 22 tanesi cinsiyet dışı otozoma veya kromozoma, ve final çifti ise cinsiyet çiftine karşılık gelir.
Cinsiyet kromozomlarının biri babadan, biri anneden gelir ve bebeğin genetik cinsiyetini belirler.
- İki X kromozomu olması (46 XX) cinsiyetin dişi olacağını belirler. Böylece birincil olarak kadın ve ikincil bir cinsiyet özellikleri gelişir.
- Bir Y kromozomu olması (46 XY) cinsiyetin erkek olacağını belirler. Böylece birincil olarak eril ve ikincil bir cinsiyet özellikleri gelişir.
Turner Sendromunda ne olur?
Bu kromozom bozukluğunu açıklanmadan önce, üreme hücresinin bölünmesinin (gametlerin) belirli yönlerini hatırlamak önemlidir.
Bu süreç mayoz olarak bilinir, mayoz I ve mayoz II olan 2 kısımdan oluşur. Bu sürecin yegane amacı ata hücreden gelen yarım genetik bilgiyi taşıyan çocuk hücreler üretmektir.
- Basitçe, 23 çift kromozomu olan kök hücre ile başlanır.
- Mayoz I sonunda, her birinde 23 kromozom bulunan 2 çocuk hücre oluşur.
- Mayoz II sonunda, bu iki hücre tekrar 23 kromozomlu 2 hücre daha üretir.
- Sonuç olarak, bir diploid kök hücre dört tane haploid çocuk hücre üretir.
Bir hücre nasıl kromozom kaybeder, ve bu durumda ne olur?
Genel olarak kabul gören teori, ikinci X veya Y kromozomunun gebelikten sonra kaybolduğudur. Yani, hücre bölümündeki bir hatadan kaynaklanan bir sorundur.
Y Kromozomunun olması dişi cinsiyet özelliklerinin gelişmesini belirler. Orijinal hücre XY olsa bile, bu kayıp dişinin gelişmesine sebep olur.
Turner Sendromunun klinik belirtileri
Fiziksel görünüş
- Çocukluk döneminde büyümede gecikme ve yetişkinlik döneminde boy kısalığı.
- Düzleştirilmiş burun kemeri, alçak göz çukurları ve göz kapaklarının hafif kusurlu olması gibi “Sfenks” benzeri yüz özellikleri.
- Normalden kısa ve geniş ense, çok alçak saç çizgisi.
- Çok sayıda renkli ben.
- Geniş göğüs kafesi, az gelişmiş çocuksu görünüme sahip ayrık meme uçları.
- İkincil cinsiyet özellikleri çok az gelişmiştir ve cinsel çocuksuluk vardır. Memeler gelişmemiştir, cinsel bölgelerde kıllanma olmaz ve yuvarlak kalçaya sahiptirler. Bütün bunlar, östrojen eksikliğinden kaynaklanır.
- Kolların dışarı dönük olması gibi kemik kusurları çok yaygındır.
Bu makaleyi ziyaret edin: Güçlü Kadınlara Özgü Nitelikler
İç bozukluklar
Ne yazık ki bu durum birtakım iç bozuklukları da beraberinde getirir, bunlar:
- Ana atardamar daralması ve kalp kapakçığı bozuklukları gibi doğuştan olan doğuştan gelen kalp rahatsızlıkları çok yaygındır.
- Önemli sayıda hastada böbrek sorunları ortaya çıkar, bunlardan en yaygını at nalı şeklinde böbrektir.
- Gadonal farklılaşma. Bu durumda yumurtalıklar az gelişmiştir, ve bunlar yerine gadonal bantlar vardır. Bu durum hipergonadotropik hipogonadizme sebep olur, yani östrojen üretimi için gerekli bütün uyarıcılar olmasına rağmen, yumurtalıklar olmadığı için üretilemezler.
- Amenore (Menstural döngünün olmaması) ve kısırlık. hastaların sadece küçük bir yüzdesi çocuk sahibi olabilir. Bu Turner Sendromunun en yaygın özelliklerinden birisidir.
Teşhis
Turner Sendromunun teşhis edilmesi zor olabilir. Bu bozukluğun olduğu çoğu kadın belirgin özelliklere sahip değildir. Hafif dışyapıya sahiptirler, çünkü daha ciddi vakalar düşükle sonuçlanır.
Vakaların üçte biri bebekken teşhis edilir. Belirli fiziksel özellikler ve kalp hastalığı belirtileri şüphe uyandıran belirtilerdir. Diğer üçte bir ise kısa boylarından dolayı çocukken teşhis edilir.
Son olarak, son üçte birlik kısım ise büyüme çağında teşhis edilir. Bu durumlarda, cinsel çocuksuluk ile birlikte kısa boy açıkça gözlemlenebilir ve hasta asla ergenliğe erişemez.
Tedavi
Östrojen kullanarak yapılan hormon replasmanı tedavisi (Hormone replacement therapy) cinsel özelliklerin gelişmesine yardımcı olur fakat kısırlığı tedavi edemez. Çocukluk döneminde, kız çocuklarında boy uzunluğunu normal seviyelere getirebilmek için büyüme hormonuyla tedavi tavsiye edilir.
Çocuk sahibi olabilirler mi?
Evet, Turner Sendromuna sahip kadınların çocuk sahibi olmaları mümkündür. Turner Sendromuna sahip insanların küçük bir yüzdesi çocuk sahibi olabilir ve gebe kalmaları için bir problem yoktur.
En sık rastlanan durum olan kısırlıkta ise, bağışlanmış yumurtalar ile in-vitro fertilizasyon (tüp bebek) tekniği kullanılmaktadır. Yumurta yapay ortamda döllenerek hastaya aşılanır. Bu gebelikler normal gebeliklere göre çok daha sıkı kontrol gerektirmektedir.
Tüm alıntı yapılan kaynaklar, kalitelerini, güvenilirliklerini, güncelliklerini ve geçerliliklerini sağlamak için ekibimiz tarafından derinlemesine incelendi. Bu makalenin bibliyografisi güvenilir ve akademik veya bilimsel doğruluğa sahip olarak kabul edildi.
- Wyss D, DeLozier CD, Daniell J, Engel E. Structural anomalies of the X chromosome: personal observation and review of non-mosaic cases. Clin Genet. 2009; 21(2):145-59.
- Jacobs PA, Betts PR, Cockwell AE, Crolla JA, Mackenzie MJ, Robinson DO, et al. A cytogenetic and molecular reappraisal of a series of patients with Turner’s syndrome. Ann Hum Genet. 1999; 54(Pt 3):209-23.
- Lippe BM. Turner’s syndrome. In: Sperling M. Pediatric endocrinology. 3 ed. Philadelphia: Saunders; 2008.p. 387.
- Chagoyén Méndez, Esther María, Álvarez Montero, José Agustín, & Zúñiga Vaca, Carmen Isabel. (2017). Síndrome de Turner en una adolescente. MEDISAN, 21(6), 720-724. scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1029-30192017000600012&lng=es&tlng=es.
Bu metin yalnızca bilgilendirme amaçlı sunulmuştur ve bir profesyonelle görüşmeyi yerine geçmez. Şüpheleriniz varsa, uzmanınıza danışın.